Reflections from building an AI‑ready experimental pipeline
OpenBind was created to address a bottleneck in structure-based AI: data. We believe that the next generation of structure-based machine learning methods requires better experimental data, not just new architectures. To address this, we are generating dense, high-quality protein–ligand datasets that link structures with binding measurements at a scale rarely available in public resources, supporting model training, fine-tuning, benchmarking, and error analysis.
Today, we are releasing the first public dataset from the OpenBind consortium: a structure–affinity dataset containing 925 crystallographic binding events from 699 compounds, and associated affinity measurements for 601 compounds.
This initial dataset focuses on a target from a major public health threat: Enterovirus A71 (EV-A71) 2A protease. By enriching the available data around this system, the release aims to substantially improve the modelling of EV-A71 2A protease and related viral proteins that are essential components of human pathogens. This target was selected in coordination with the AI-driven Structure-enabled Antiviral Platform (ASAP) Discovery Consortium, a global antiviral discovery center for pandemic preparedness focused on delivering therapeutics for globally equitable and affordable access.
Alongside the experimental data, we provide reference benchmarks for conventional docking, machine-learning-based docking, cofolding, and affinity prediction methods for the community to build upon.
This represents the first public demonstration of the data generation infrastructure we have built to enable the next generation of structure-based AI. It illustrates the nature of experimental data that OpenBind is now poised to generate rapidly and repeatedly across numerous future targets.
Data: Zenodo / Fragalysis
Benchmarks: GitHub
Experimental protocols: OpenBind protocols.io Workspace
Further information about OpenBind: https://openbind.uk/
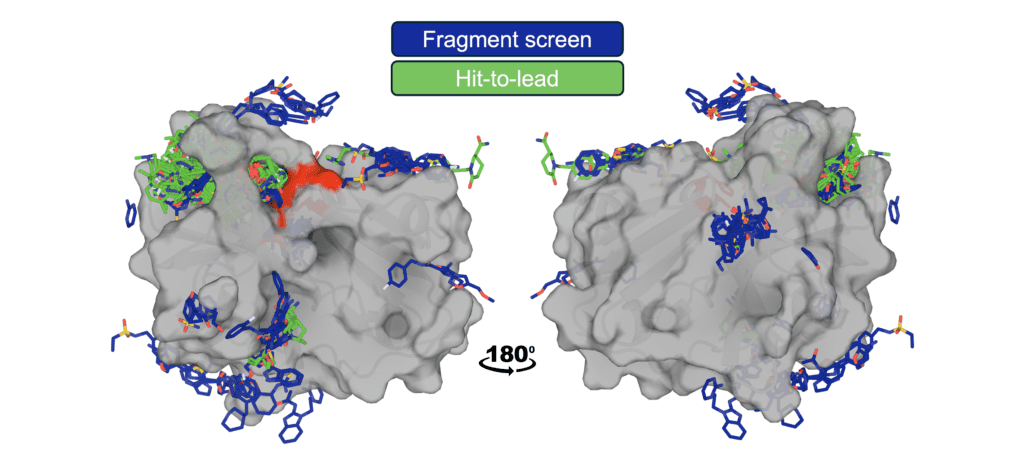
Figure: Structural coverage of ligand binding to EV-A71 2A protease. EV-A71 2A protease is shown with ligands from the OpenBind release overlaid, illustrating the density of experimentally observed binding modes generated through the fragment screen and follow-on compounds. The catalytic triad is highlighted in red.
An invitation to the community
We hope this release becomes a useful resource and testbed for the community. Use it to train, fine-tune, and stress-test models; to evaluate docking, cofolding, and affinity prediction methods; to study receptor choice and pose confidence; and to ask whether models capture local structure–activity relationships across related compounds.
We are especially interested in how current approaches fail. If the dataset exposes a weakness in a method, a benchmark, or an assumption in the field, that is valuable. OpenBind will improve through community use, critique, and participation.
Most importantly, tell us what you learn.
OpenBind is generating the experimental data AI needs to propel drug discovery.
OpenBind is an open-science initiative generating large-scale, high-quality experimental protein–ligand data for structure-based AI to deliver new biological insights and accelerate drug discovery. The first phase of OpenBind focused on building the practical infrastructure needed to do this repeatedly and with high velocity: target preparation, crystallographic workflows, compound progression, affinity measurement, data processing, benchmarking, and release mechanisms. This release marks the first public output from that process.
Our aim is not simply to release more structures, but to generate the most informative data for AI methods in structure-based drug discovery. OpenBind is designed to generate data where the learning signal is high: targets and compound series that are under-represented in public resources, challenging for current methods, and valuable for expanding the capabilities of structure-based AI.
This is made possible by combining Diamond Light Source’s world-leading high-throughput experimentation capabilities with state-of-the-art computational modelling. These data are intended to support training, fine-tuning, benchmarking, blind challenges, and the development of new methods for docking, cofolding, affinity prediction, active learning, and molecular design.
The first OpenBind dataset is a dense structure–affinity dataset for EV-A71 2A protease
The enteroviral 2A protease is a virally-encoded cysteine protease that supports enterovirus replication by processing the viral polyprotein and cleaving key host factors involved in translation and antiviral signalling. Its essential role in the viral life cycle, combined with a druggable protease active site, makes it an important target for structure-guided ligand discovery. EV-A71 2A protease is also closely related to 2A proteases from other picornaviruses that are pathogenic to humans, so improved methods for modelling these systems could have broader relevance for antiviral discovery.
The release contains 925 crystallographic binding events from 699 compounds, generated from a crystallographic fragment screen and subsequent follow-on compounds. For 601 of these compounds, we are also releasing associated affinity measurements, reported as KD values measured using the Creoptix WAVEsystem, linking structure to strength of binding. All data were generated using Coxsackievirus A16 (CVA16) 2A protease as a surrogate system for EV-A71 2A protease. These two proteins differ in only five positions in the amino acid sequence, none of which are close to the active site.
This is not just a collection of isolated protein–ligand complexes. It links many related compounds, crystallographic binding modes, and affinity measurements. This makes the dataset useful for training, fine-tuning, benchmarking, and error analysis: users can ask whether predicted poses match observed binding modes; whether related compounds preserve, shift, or lose interactions; whether affinity models capture local structure–activity relationships; how docking and cofolding methods behave across a consistent experimental series; and whether fragment-screen data can improve prediction on follow-on compounds.
We also provide reference benchmarks of current methods across docking, cofolding, and affinity prediction. These benchmarks give an initial view of how existing approaches perform on this dataset; they are not intended to define a final leaderboard, and we hope the community will improve on them, critique them, and extend them.
Complete experimental protocols can be found in the OpenBind protocols.io Workspace
What does the first OpenBind dataset add beyond existing public data?
A natural question for any new structure–affinity release is whether it adds genuinely new information beyond existing public data. This matters for both training and evaluation. If a benchmark is very close to protein–ligand complexes already present in public datasets, it can be difficult to tell whether a method is generalising to a new system or benefiting from near-neighbour information. This issue has become especially important for machine learning methods. Runs N’ Poses showed that the performance of cofolding models depends strongly on the similarity of the test data to the training data.
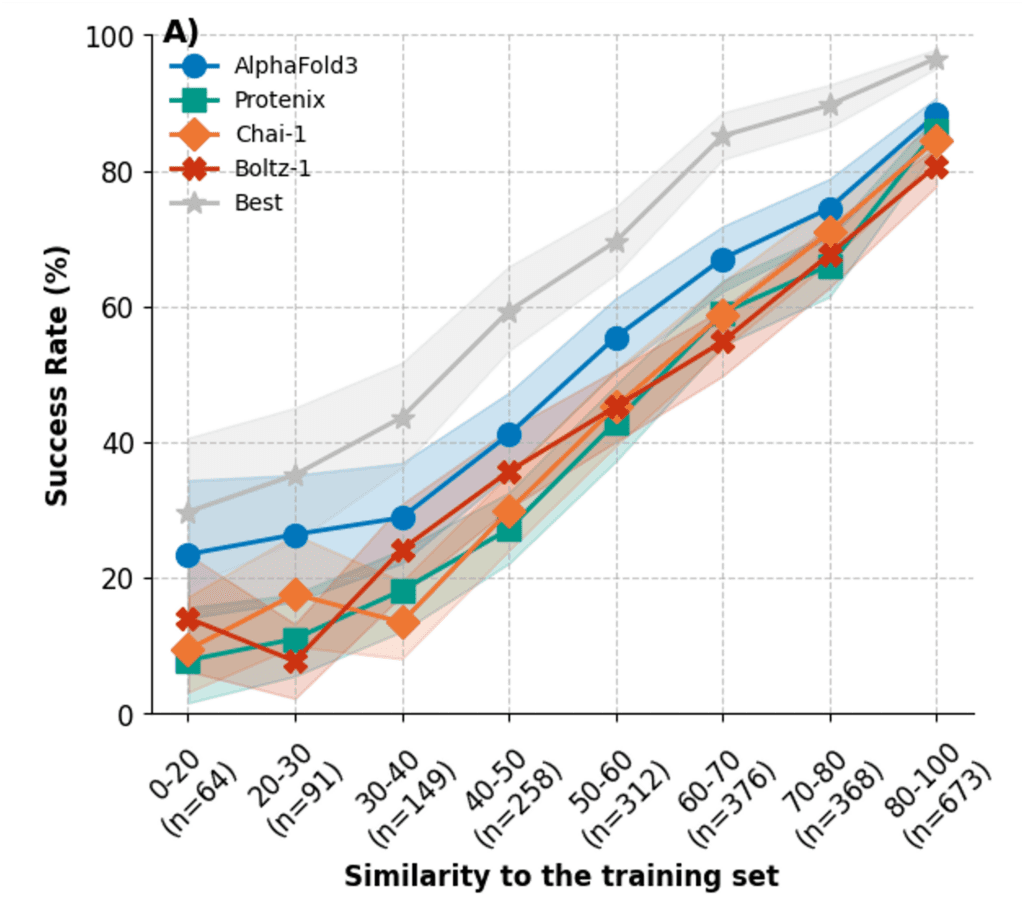
Figure: The success rate of cofolding methods in predicting protein–ligand complexes on the Runs N’ Poses benchmark drops off dramatically as binding interface similarity to data in the training set decreases. [Figure from https://doi.org/10.1101/2025.02.03.636309]
We therefore used the same ligand–pocket similarity metric to compare the EV-A71 2A protease complexes with public protein–ligand structures released before 30 September 2021, the structural training cutoff for AlphaFold3 and many other cofolding models.
The EV-A71 2A protease complexes are relatively dissimilar to existing PDB protein–ligand data under this metric. This OpenBind dataset therefore provides a substantial experimental dataset for testing docking, cofolding, and affinity-prediction methods, helping address a central question for structure-based AI: how well do current methods perform on a coherent, experimentally-determined target dataset that is not already well covered by near-neighbour structures?
The dataset contains many related structures from a fragment screen and follow-on chemistry, with binding measurements for many compounds. This makes it useful for studying how models learn within a chemical series, how pose and affinity predictions respond to local chemical changes, and where current methods fail.
This similarity analysis should not be overinterpreted. Similarity to existing public data is only one aspect of benchmark difficulty; ligand chemistry, binding-site flexibility, assay coverage, task definition, and evaluation protocol all matter. But it provides an important sanity check: this release adds a dense structure–affinity dataset in a region of protein–ligand space that appears relatively under-represented in existing public structural data.
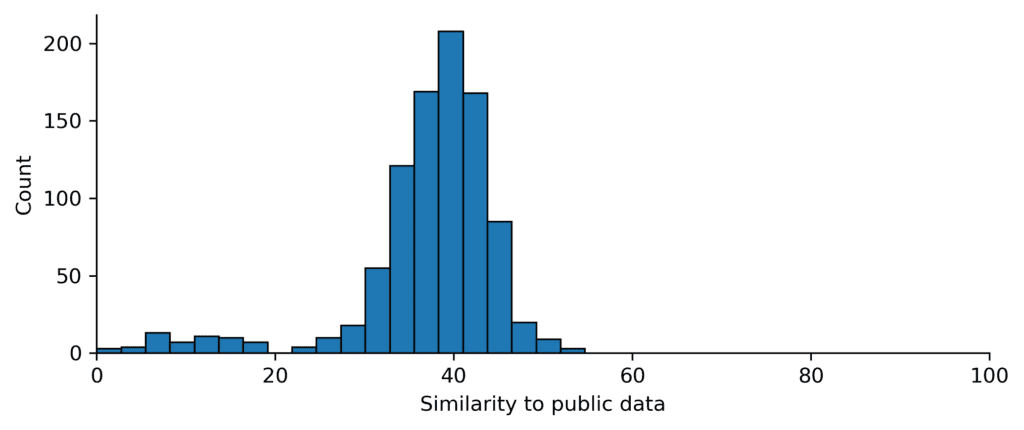
Figure: The OpenBind EV-A71 2A protease complexes are dissimilar to existing public data. Similarity of EV-A71 2A protease complexes to existing public protein–ligand structures available before the AlphaFold3 training cutoff date (30 September 2021), measured using the similarity metric proposed in Runs N’ Poses (“sucos_shape_pocket_qcov”).
How well do current-generation models perform?
Alongside the first OpenBind data package, we are releasing initial reference evaluations across several families of structure-based modelling methods – conventional docking, machine-learning-based docking, cofolding, and affinity prediction – to see how well they perform prospectively on new protein–ligand data.
We tested established docking tools such as AutoDock Vina, ML-based docking approaches such as GNINA and DiffDock, recent cofolding methods including Boltz, OpenFold3, and AlphaFold3, and several affinity-prediction methods.
The purpose of these benchmarks is not to declare a “best” method. Different prediction tasks relevant to drug discovery and different experimental setups test different capabilities. Instead, these benchmarks provide an initial reference point for the community: a way to understand how current methods behave on a single-target structure–affinity dataset containing many related compounds from the same experimental campaign, which could not have been used during method development.
Here, we summarise the main results across pose prediction, complex prediction, and affinity prediction. Our analysis considered all structures that passed PoseBusters validity checks and were not suspected to be crystal contacts (n=881). Of these, 79 were fragment binders. The analysis below focuses on the follow-on compounds (n=802). We considered a prediction successful if the ligand was within 2 Å heavy-atom RMSD of the experimental pose, the protein–ligand complex achieved an LDDT-PLI score ≥ 0.8, and the prediction passed PoseBusters validity checks. Unless otherwise stated, we report the top 25 success rate, in line with recent publications, such as Runs N’ Poses and OpenFold3.
We hope the community will improve on these results, but also use them to ask better questions: where do methods fail, which failures matter, and what kinds of evaluation best reflect real structure-based design?
Pose prediction: redocking is strong, cross-docking is harder
We evaluated conventional and machine-learning-based docking methods on their ability to recover crystallographic ligand poses.
Model performance strongly depended on the task. In redocking, where the cognate protein structure is provided, the best performing methods achieve high success rates. For example, GNINA achieves an success rate of 85% across the 802 binding events of the follow-on compounds. This shows that, when the receptor is already in a ligand-compatible conformation, current docking approaches can often recover the observed binding mode the majority of complexes.
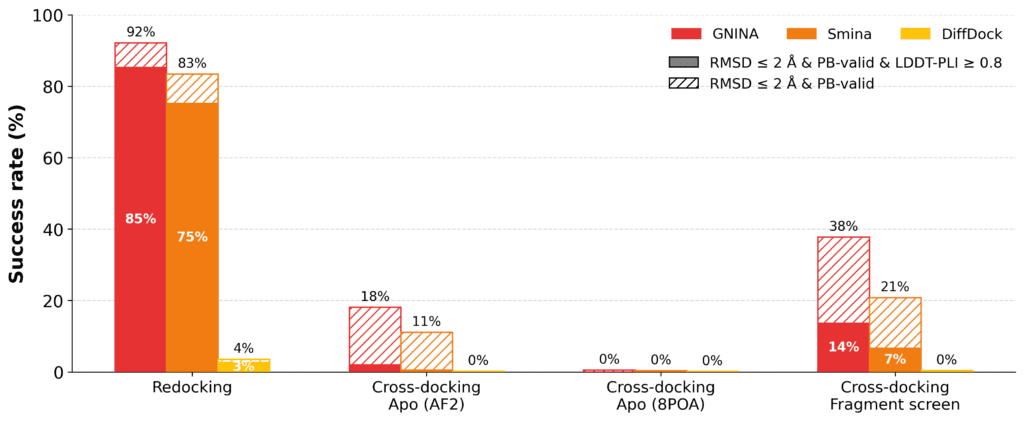
Figure: Docking performance on the EV-A71 2A protease complexes. Redocking performance is strong, while cross-docking into apo structures is substantially more challenging. Using a receptor structure from the most similar fragment improves pose recovery, but success rates remain well below redocking, highlighting the importance of receptor conformation.
Cross-docking is substantially more challenging. We first docked ligands into an experimentally-determined apo structure (PDB ID: 8POA) and the predicted apo structure from the AlphaFold DB. In both cases, the success rates drop to less than 5% for all methods. This is caused by a conformational change in a loop forming part of the binding site: in the apo and AF2 structures, this loop adopts a distinct conformation from most ligand-bound structures, partially occluding the ligand-binding pocket and preventing recovery of the crystallographic binding mode without steric clashes.
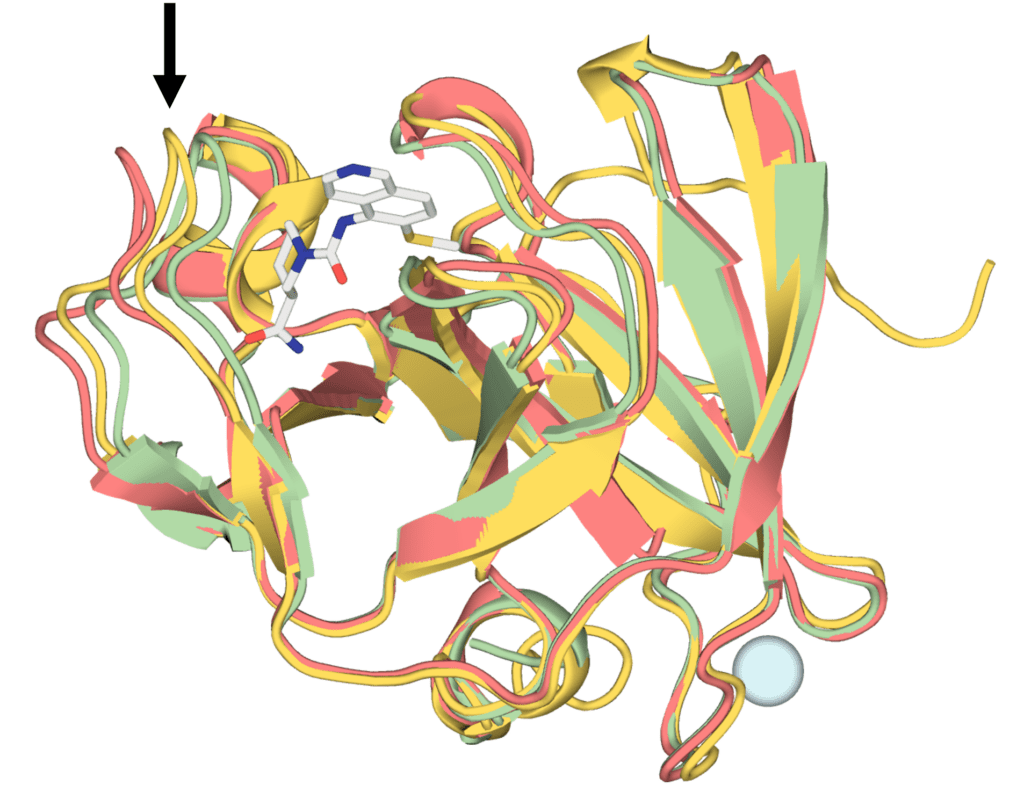
Figure: Comparison of the protein structures for ligand-bound structure x5261a (red), experimentally determined apo structure (PDB ID: 8POA, green), and AF2-predicted apo structure (yellow). Different loop configurations in the three structures (indicated by the arrow) prevent successful rigid-body cross docking in almost all cases.
We therefore also docked each non-fragment compound into the protein structure from the fragment with the greatest chemical similarity, measured by Tanimoto similarity of ECFP4 fingerprints. While a rudimentary method for selecting a protein structure for cross-docking, this substantially improved pose recovery. For example, GNINA placed 38% of compounds within 2Å RMSD of the crystallographic pose, although only 14% satisfied the stricter joint criterion of RMSD ≤ 2Å and LDDT-PLI ≥ 0.8. This suggests that choosing a receptor conformation from a chemically related ligand can recover some of the lost performance, but that reproducing the detailed protein–ligand interaction pattern remains more difficult.
This distinction is important. Redocking tests whether a method can reproduce a pose when the correct receptor conformation is already available. Cross-docking tests something closer to many practical structure-based workflows: whether a method can handle receptor conformational mismatch. For the EV-A71 2A protease complexes, that mismatch is often the dominant factor in whether a pose can be recovered. The dataset therefore provides a useful test case for separating failures in ligand placement and scoring from failures in receptor-state selection, and for evaluating whether ensemble docking, receptor relaxation, induced-fit approaches, or learned docking methods can overcome this conformational challenge.
Cofolding substantially outperforms cross-docking by reducing receptor mismatch, but remains below redocking
The cross-docking results highlight a central difficulty in structure-based modelling: if the binding site is in the wrong conformation, it may be impossible to recover the crystallographic pose.
Cofolding offers an alternative route to this problem. Rather than docking a ligand into a fixed receptor structure, these methods predict the protein–ligand complex directly, allowing the binding site conformation and ligand pose to be modelled together. We evaluated recent cofolding methods using the same success criterion as above.
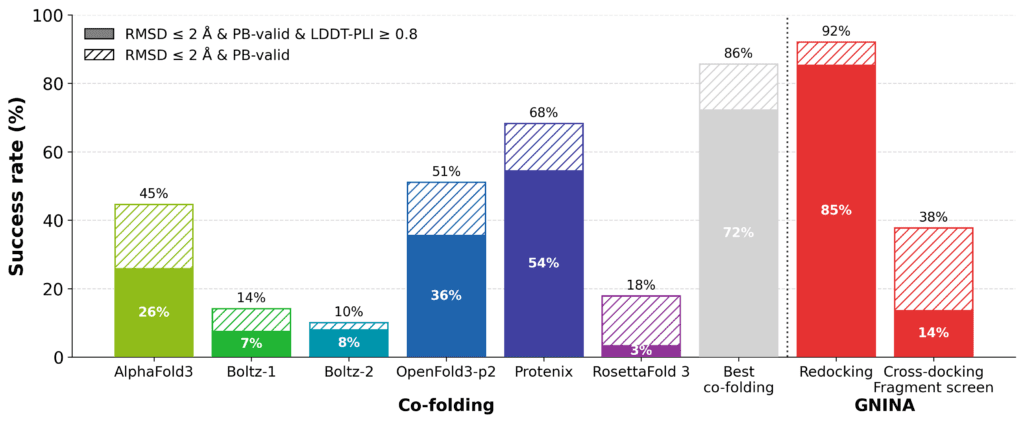
Figure: Cofolding performance on the EV-A71 2A protease complexes. Cofolding methods substantially outperform fixed-receptor cross-docking on this dataset, but remain below the success rate of redocking.
Overall, cofolding improves success rates compared to cross-docking , but remains well below redocking performance, where the cognate receptor structure is provided. There is considerable variation between methods. Protenix and OpenFold3-p2 achieve the strongest individual performance among the methods tested, while AlphaFold3, Boltz-1, Boltz-2, and RF3 show lower success rates on this dataset. An oracle “best of cofolding methods” reaches substantially higher success rates than any single method, suggesting that different methods succeed on different subsets of complexes, albeit this upper limit is calculated across significantly more predicted structures. This highlights an opportunity for better methods for ranking predicted complexes.
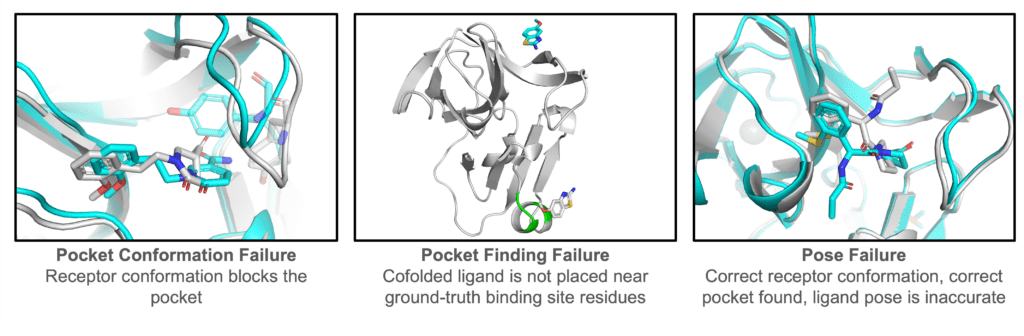
Figure: Representative failure modes. Aggregate metrics are useful, but they do not tell the whole story. Here are three interesting classes of failure of cofolding methods.
Fragment screen data significantly improves cofolding performance
One goal of OpenBind is not only to evaluate existing methods, but to provide the experimental data needed to improve them. The EV-A71 2A protease release is well-suited to answer this question because it contains both fragment-bound structures and follow-on compounds from the same experimental campaign. This makes it possible to ask whether early fragment-screen data can provide a useful learning signal for predicting the follow-on compounds.
As an initial test, we fine-tuned OpenFold3-p2 using the fragment-bound structures from the crystallographic screen and evaluated performance on the follow-on compounds.
Fine-tuning on fragment-screen data substantially improved cofolding performance. The success rate of OpenFold3-p2 increases from 36% to 76%, approaching that of redocking with GNINA (85%). This is significant, since redocking is provided with the cognate receptor structure, whereas the fine-tuned cofolding model must still predict the protein–ligand complex. At the same time, we did not train or adapt redocking methods on the target-specific fragment data, which would likely notably improve docking performance.
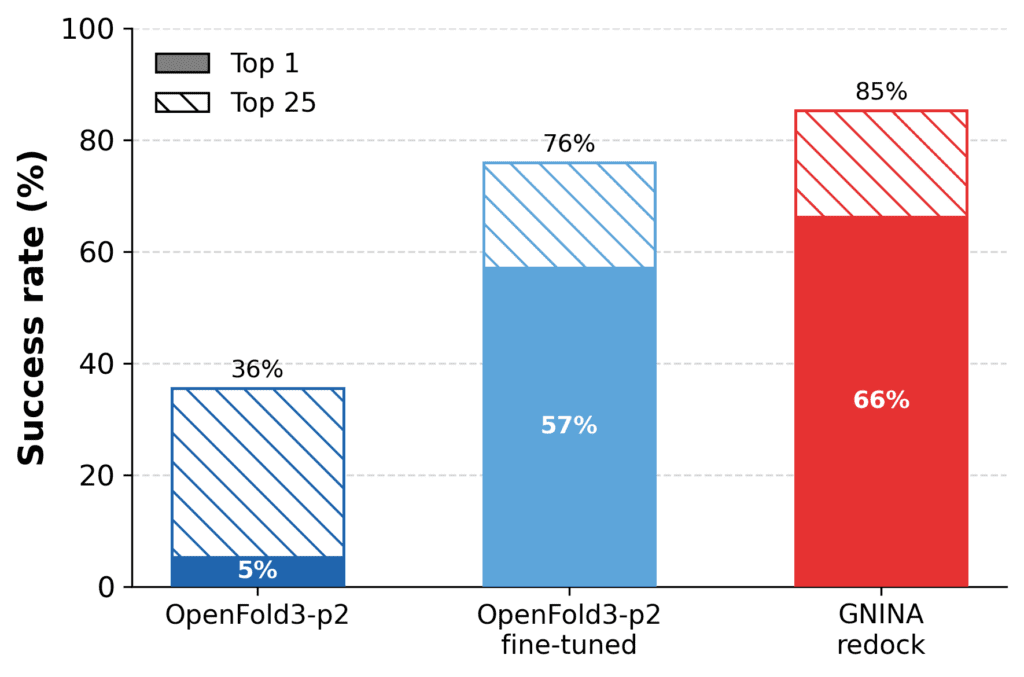
Figure: Fragment-screen fine-tuning improves cofolding performance. Cofolding performance before and after fine-tuning on fragment-bound EV-A71 2A protease structures, evaluated on held-out follow-on compounds, defined as RMSD ≤ 2Å, LDDT-PLI ≥ 0.8, and PoseBusters valid. Fine-tuning substantially improves success rates and approaches, but does not fully reach redocking performance.
The result suggests that fragment-screen structures can provide more than starting points for medicinal chemistry. They can also provide useful training data for structure-based AI, helping models learn target-specific binding-site geometry, ligand interactions, and receptor conformational preferences. This is exactly the kind of learning signal OpenBind is designed to generate.
Affinity prediction remains challenging: Simple baselines are hard to beat
The release contains affinity measurements for 601 compounds from the Creoptix WAVEsystem, corresponding to 804 of the 925 crystallographic binding events. For the affinity-prediction benchmark, we applied quality-control filters to the biophysical measurements and retained 494 compounds for evaluation. We then evaluated several affinity-prediction methods on their ability to predict or rank binding strength, comparing structure-based models with simple physicochemical baselines based on molecular weight and cLogP.
Affinity prediction remains difficult on this dataset. Several methods produce only modest correlations with measured affinity. Boltz-2 and GNINA show the strongest correlations among the model-based methods, while AEV-PLIG and Boltz-2 give the lowest RMSE. However, the simple molecular-weight baseline achieves the highest Spearman correlation overall.
This result should not be interpreted as meaning that molecular weight is a generally sufficient model of binding affinity. Rather, it is a reminder that affinity prediction benchmarks must include simple baselines and must be interpreted carefully, especially within a single experimental campaign where potency may correlate with compound elaboration, size, or series progression. A method that uses structural information should be expected to outperform such baselines if it is capturing the protein–ligand interactions that drive affinity.
For OpenBind, this is an important result. The structures and affinity measurements in this release provide a realistic testbed for asking whether affinity models can learn more than coarse chemical trends: can they capture local structure–activity relationships, distinguish productive from non-productive elaborations, and connect observed binding modes to measured binding strength?
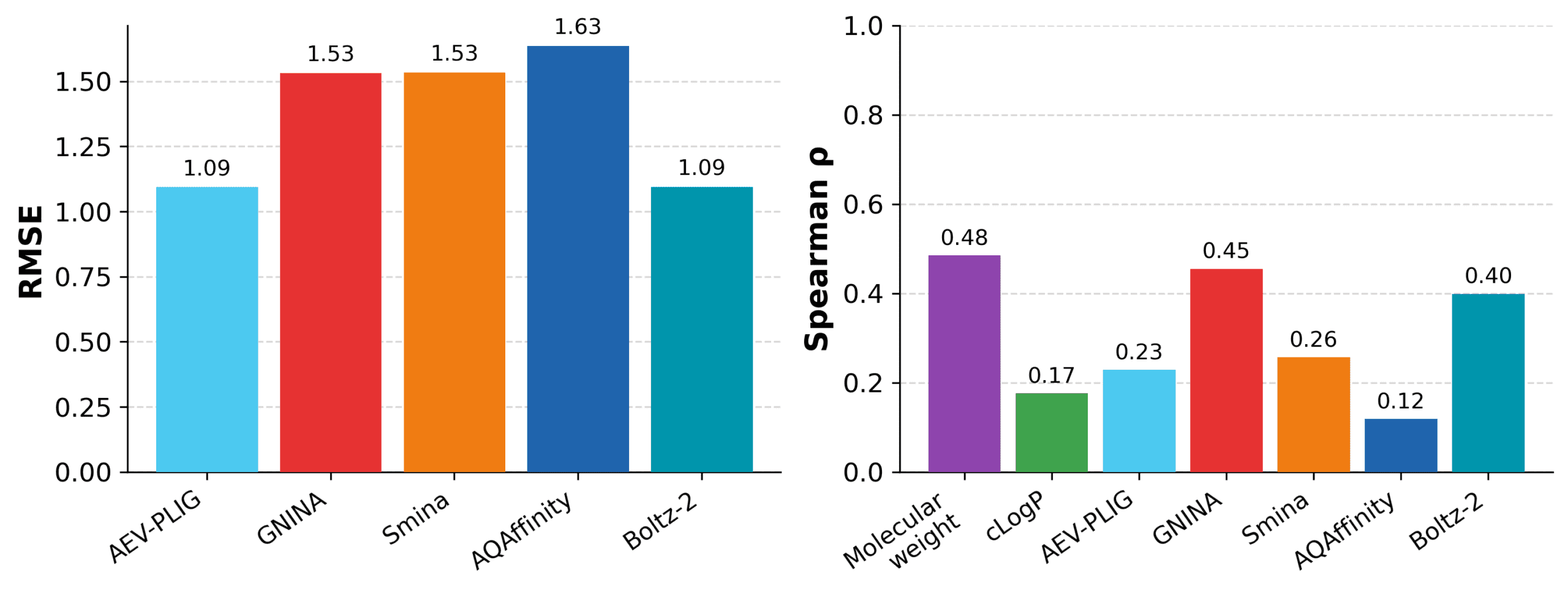
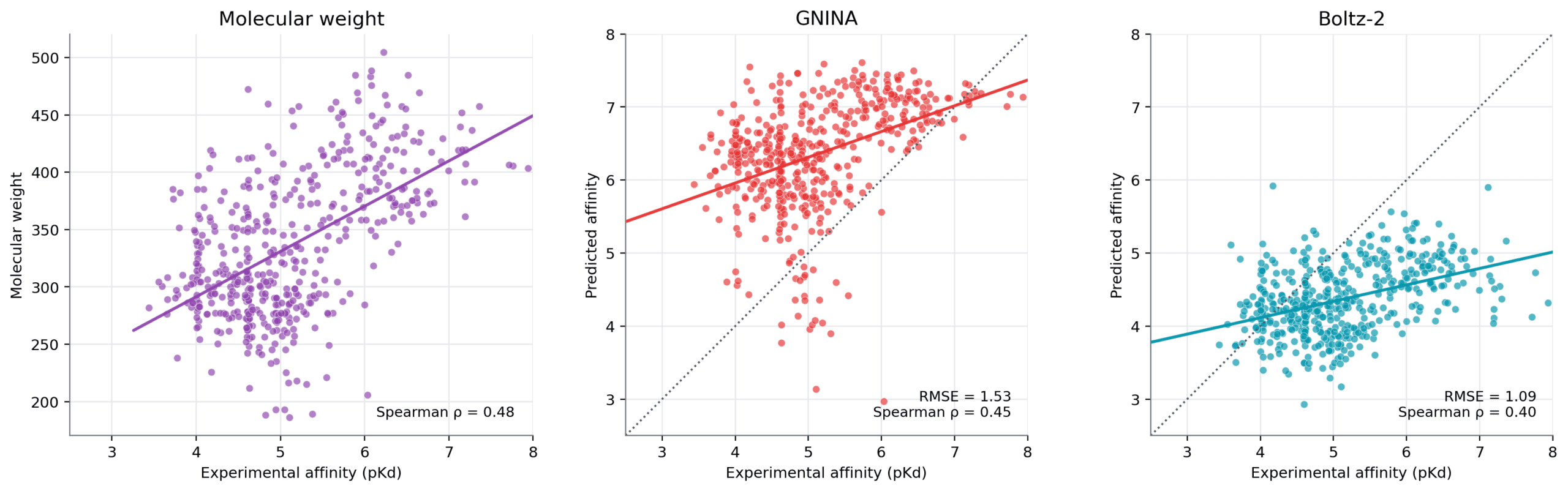
Figure Affinity prediction remains challenging for structure-based prediction methods. across the EV-A71 2A protease series. Molecular weight provides the strongest correlation baseline, while Boltz-2 and GNINA show the strongest correlations among the structure-based methods tested.
Caveats and interpretation
EV-A71 2A should be interpreted as real experimental campaign data, not as a perfectly balanced benchmark.
Not every compound has every type of data: the release contains over 925 crystallographic binding events from 699 compounds and 601 affinity measurements, but the structure and affinity coverage do not match one-to-one. The data are also not uniformly distributed across chemical space. Some regions are more densely explored than others, reflecting the fragment screen, follow-on chemistry, experimental feasibility, and the decisions made during the campaign.
The benchmark results should be viewed in the same spirit. Redocking, cross-docking, cofolding, and affinity prediction test different capabilities and should not be collapsed into a single notion of “best method.” The reference results depend on specific protocols, inputs, and evaluation choices. We expect users to improve on them, critique them, and propose alternatives.
We see these caveats as part of the value of the data release. Structure-based discovery is not a clean benchmark problem. It is shaped by incomplete measurements, local chemical series, receptor conformations, experimental constraints, and sequential decisions. Benchmarks that remove all that complexity are testing the wrong thing.
The EV-A71 2A dataset captures many of the features that make real structure-based discovery campaigns scientifically valuable and computationally challenging. This release therefore serves two purposes: it provides a substantial dataset for one antiviral target system, and it demonstrates a data-generation model that OpenBind is poised to repeat across future targets.
Data: Zenodo / Fragalysis
Benchmarks: GitHub
Experimental protocols: OpenBind protocols.io Workspace
Further information about OpenBind: https://openbind.uk/
Data license: CC0 1.0 Universal
DOI: 10.5281/zenodo.20026661
Want to learn more, raise issues, or ask questions? Join the OpenBind Data Hub Discord server
Acknowledgements
We thank everyone who was involved in generating and processing data, and preparing this release, as well as the funders supporting OpenBind.
OpenBind received funding from the UK Department of Science, Innovation and Technology under grant number G2-SCH-2025-06-16537.



