Structure‑based AI is hungry for data – good data – and there simply hasn’t been enough of it. OpenBind aims to fix that by generating high‑quality experimental protein–ligand datasets at a scale rarely seen in the public domain.
By pairing structural information with binding measurements, OpenBind provides the foundation needed for training and evaluating the next generation of AI models in biology and drug discovery. This release is the first glimpse into that growing dataset.
In this post, we provide more general information about how the data from our first OpenBind data release package was generated, containing 601 compounds, of which 494 were used in our analysis after QC checks.
Where to find the data
Experimental Data Download:
- This Fragalysis-download-link will download about 600Mb of data, which contain the structures (aligned as described here) as well as affinities (look inside the extra_files_1 subdirectory). Further documentation for navigating the downloaded data can be found in Fragalysis-read-the-docs.
- Try this Fragalysis-data-view-link for a direct 3D view, which is fully documented here
- Detailed experimental protocols can be found on the OpenBind protocols.io Workspace
ML Data Download and Code:
Licencing and other useful links:
Data license: CC0 1.0 Universal
Background on the protein target
Picornaviridae, such as Enterovirus A71 and Coxsackievirus, are the causative agents for hand, foot and mouth disease, affecting mostly infants and children. However, on rare occasions, these viruses can also cause more severe neurological symptoms. Currently, no vaccine or treatment is available.
The 2A cysteine protease (2APro) is essential for viral replication and performs an intramolecular cleavage to release the VP1 portion of the viral polyprotein. Inhibition of this step is anticipated to yield sub-stoichiometric inhibition of viral growth, making it an attractive target. This enzyme has also been implicated in the suppression of the host immune response by cleaving host factors.
How it all came together
This first OpenBind data release builds on earlier research efforts aimed at developing pan‑enteroviral 2A protease inhibitors, undertaken by the AI‑driven Structure‑enabled Antiviral Platform (ASAP) Discovery Consortium.
The fragment screen against Coxsackievirus A16 (CVA16) 2A protease, which is used as a surrogate system for Enterovirus A71 (EV-A71), was done in 2024. The hit-to-lead programme under the ASAP Discovery Consortium, with structural efforts by Diamond Light Source, allowed the OpenBind Consortium to now take advantage of the availability of 2APro binders with a range of potencies and enrich the available data around this target – aiming to enhance modelling performance across a wider set of related proteins that play essential roles in major human pathogens.
To be able to release a data package of 601 compounds, of which 494 were used in our analysis after QC checks, required a massive effort: More than 4,300 fragments and follow-up compounds had to be soaked, with data collected on over 7,600 crystals. Even though the data from this number of crystals could technically be collected in ‘just’ two weeks, all datasets were still manually built and refined, which fellow crystallographers know takes weeks to months. In the future, the process will need less human intervention, as we were simultaneously working towards more automated ligand-fitting and model-building pipelines during the first Phase of OpenBind.
Affinity Dataset
Developing the Creoptix Protocol for 2APro
High-quality affinity data starts long before the first compound is injected. For this first data release, OpenBind invested up front in a Design-of-Experiments (DoE) campaign to develop a Creoptix grating-coupled interferometry (GCI) protocol that would outperform a standard buffer condition (i.e. HBS-P) based assay on the three things that, in our experience, matter for downstream modelling: binding response, on-chip stability, and sensorgram quality. The result is the protocol underpinning every KD value in this release and, just as importantly, a protocol that is well-enough understood that our production run completed without surprises.
A Three‑Stage Design‑of‑Experiments (DoE) Funnel
Our protocol-development workflow is a triage funnel that escalates from cheap, high-throughput stability measurements in solution to long-running on-chip experiments that mimic a full production day (Fig. 1).
- NanoDSF (protein stability in solution): A typical screen runs ~224 conditions across roughly two days of runtime and analysis, and tells us which buffer families are worth taking onto a chip.
- Creoptix buffer screening (protein stability, signal quality, and binding response on a control sample): A typical screen is ~24 conditions, ~2 days of instrument time, and historically ~4 days of analysis and visual inspection; this can conceivably be collapsed to ~1 day with an auto-analysis pipeline.
- Stability test (long-injection performance vs. the standard method): A typical experiment is ~5 conditions run for 20 h each, with analysis turning around in ~2 days and expected to reduce to at least 1 day with the auto-analysis workflow.

Figure 1: The OpenBind Creoptix DoE funnel; (a) nanoDSF buffer triage feeds into (b) Creoptix buffer screen, which in turn feeds (c) long-injection stability test that benchmarks the candidate protocol against the standard HBS-P condition method.
Immobilisation, buffer, detergent, and weak-binder sensitivity: what the DoE locked in
Four checkpoints set the backbone of the protocol. First, immobilisation chemistry: amine coupling killed 2A-protease activity entirely (no binding response or negative response), while streptavidin-biotin capture of the biotinylated protease recovered a clean, positive binding response.
Second, buffer pH: matched pH 7 vs. pH 8 comparisons consistently favoured the lower pH on maximising reference-subtracted binding response.
Third, detergent (held against 10 mM HEPES pH 7, 100 mM NaCl, 0.5 mM TCEP, 2% DMSO): no detergent gave high non-specific interaction with reference surface, CHAPS gave variable profiles between repeats, and LMNG, DDM, and Tween-20 produced acceptable, well-shaped sensorgrams with reproducible association and dissociation phases.
Fourth, weak-binder sensitivity: testing two IC50 > 90 µM samples alongside three stronger controls across Tween-20, DDM, and LMNG, the weak binders gave no usable signal in Tween-20 (in fact negative responses) but produced clear binding signal in both DDM and LMNG, while the stronger controls were detected across all three detergents, although the standard Creoptix software could not fit a kinetic or affinity model to the weak-binder DDM/LMNG sensorgrams.
That last point is the bridge to the next post in this series: the signal is there, the standard fitting tooling just cannot extract it, and our analysis post explores numerical fitting and alternative modelled sensorgrams.
Stability over a
20 h injection series
Production runs at OpenBind routinely span a full day on a single chip surface, so any candidate buffer must hold the protein active over that window. Following the buffer screening experiments and to benchmark stability, four buffer conditions were selected.
We injected a control compound hourly for 20 h and tracked the apparent KD across the series; a moving KD is a direct readout of the surface or protein degrading underneath the assay. Of the conditions tested, 10 mM HEPES pH 7.0, 100 mM NaCl, 0.005% Tween-20, 0.5 mM TCEP, 2% DMSO gave the flattest KD profile over 20 h, with stability considered most critical, this running buffer was selected for this data release.
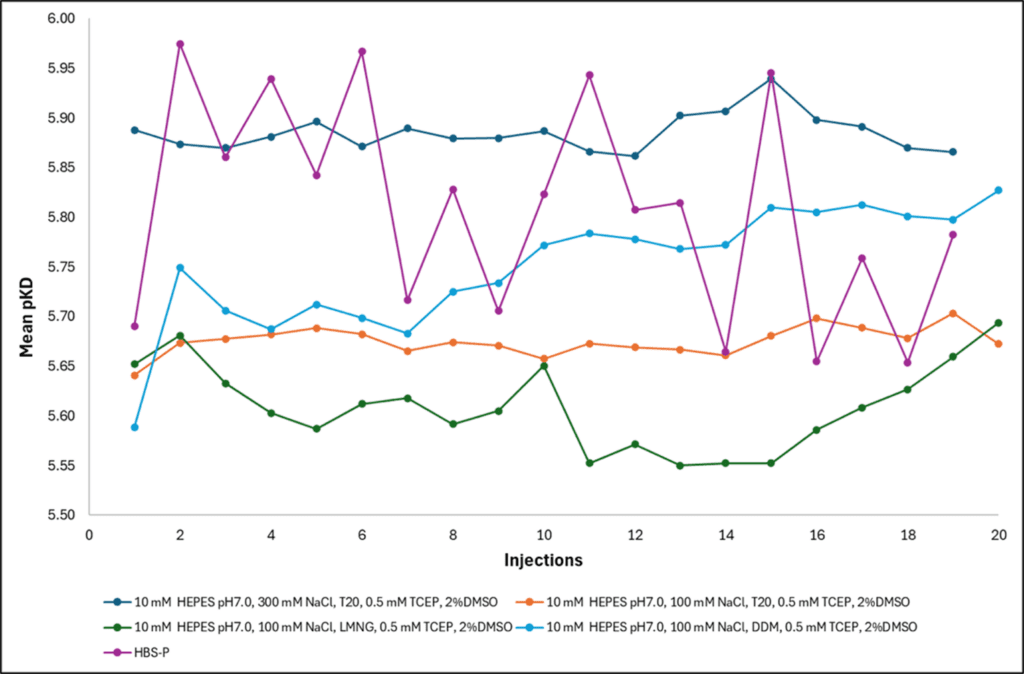
Figure 2: Apparent KD of a control compound injected hourly over 20 h across the candidate stability buffers. The 10 mM HEPES pH 7.0, 100 mM NaCl, 0.005% Tween-20, 0.5 mM TCEP, 2% DMSO condition held a consistent KD across the full series.
What this means for OpenBind’s first data release
The headline result of all of the above is mundane in the best possible way: the 2A-protease production run that generated the data in this release proceeded without hiccups; affinities of 494 compounds were successfully measured by grating-coupled interferometry (GCI) on a Creoptix WAVEsystem using waveRAPID kinetics. Over 2,000 sensorgrams were generated (from over 600 compounds in triplicate) and were manually inspected, fitted, and analysed by trained experimentalists using the vendor software.
This last step took two weeks to complete and led to the release of high-quality affinity evaluations for a total of 494 compounds. That is not luck, it is a direct consequence of OpenBind having spent the time up front to develop a robust protocol: a buffer that holds the surface stable across a full production day, a detergent that gives clean sensorgrams, and a pH that does not cost binding response. We believe that if the protocol is well characterised, troubleshooting at production time stops being archaeology and becomes a short checklist.
Open challenges, and what comes next
A KD is an abstraction. It is the number that falls out of a particular protocol, processed with a particular numerical method, fitted to a particular binding model; change any of those three, and the number moves. Our view at OpenBind is that the right question for ML-relevant affinity data is not “how close to the true KD did we get?” but “is the data good enough to resolve the activity differences that matter for downstream modelling?”
The protocol described here is the first step. Our next post in this series digs into the data-analysis side: numerical methods for extracting affinities and what the implications are for the affinity values reported in this release. To reduce human intervention needed in the affinity workflow as well, our developers and experimentalists were working together to build SensoFit, an open-source Python tool that not only processes Creoptix experiments automatically, but also offers a more transparent and accessible approach to the inspection, fitting and analysis of the measurements. If you want to know more about the data fitting, read our Data Fitting blogpost.
Structure Dataset
X-ray crystallography is a highly sensitive method for detecting even weak binding events and can be performed in a high-throughput manner, screening 1,000 compounds in a single week. The readout provides detailed insights into the binding mode and interactions of ligands with their protein targets. The Coxsackievirus A16 2A protease was used as a surrogate system for the EV-A71 2A protease. The 2A protease sequence of EV-A71 and CVA16 differs by 5 amino acids, none of which are near the catalytic site or predicted to affect protease activity. The CVA16 2A protease was expressed in E. coli BL21(DE3) as a His6-SUMO fusion protein and purified using affinity and size exclusion chromatography.3
Crystals of the 2APro were soaked with fragments from the DSi-Poised, SpotXplorer, FragLites, PepLites, York 3D, Minifrags, and Covalent MiniFrags fragment libraries at a final concentration of 50-100 mM. Follow-up compounds designed by the ASAP Discovery Consortium during the hit-to-lead programme were soaked at a nominal concentration of 2-10 mM. The crystals were harvested and cryo-cooled before diffraction data were collected at the macromolecular crystallography beamlines I03 and I04-1 at Diamond Light Source.4
Diffraction data were automatically processed using Diamond Light Source’s automated analysis pipelines, and further analysis was performed through XChemExplorer. Initial map calculation was carried out using DIMPLE. Ligand CIF restraints were generated using GRADE or GRADE2. Hit identification was performed using PanDDA2. Refinement and model building were carried out using COOT, and REFMAC or Buster via the XChemExplorer platform. 467 of the structures were previously deposited to the PDB under Group Deposition IDs G_1002288, G_1002329, G_1002330, and G_1002344, while the OpenBind data package also includes 265 new and previously unreleased structures.
Meet the OpenBind Data Production Team











Questions
If you have specific questions regarding the OpenBind data release package, we have a Discord channel to reach out to us: OpenBind Data Hub
Acknowledgements
We would especially like to thank other members of the ASAP Discovery Consortium, our OpenBind colleagues from the Protein Production Team, and our colleagues at beamline I04-1 and I03 for their support and involvement in generating this data package. Special thanks go to Ryan Lithgo, who was leading the structural biology efforts of the 2A programme in the early phases, and Grant Watt, who has made invaluable contributions to model building and refinement for this data package.
References
1. https://dx.doi.org/10.17504/protocols.io.kqdg3mn3ql25/v1
2. waveRAPID—A Robust Assay for High-Throughput Kinetic Screens with the Creoptix WAVEsystem
3. Enterovirus coxsackievirus A16 2A protease small scale expression and purification protocol V.3



